Marfan Syndrome is a genetic disorder of the connective tissue in your body. The damage caused by Marfan syndrome can be mild or severe. The most serious effects of Marfan syndrome can be life-threatening. People with Marfan syndrome are usually tall and thin, with disproportionately long arms, legs, fingers and toes. Patients sometimes also have “arachnodactyly”, which means “spider-like fingers”, since one of the characteristic signs of the disease can include very long fingers and toes.
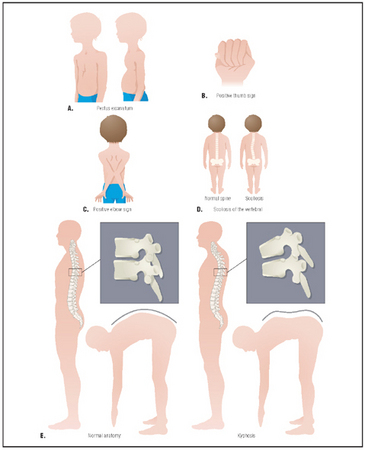
The signs of Marfan syndrome: (left to right) pectus excavatum, positive thumb sign, positive elbow sign, normal spine compared with scoliosis, normal anatomy compared with kyphosis.
Cardiac and circulatory abnormalities
About 90 percent of Marfan patients will eventually develop cardiac complications, that may include:
- Aortic enlargement. This is the most serious potential complication of Marfan syndrome. Because of the abnormalities in connective tissue, the walls of the aorta (the large blood vessel that carries blood away from the heart) are weaker than normal and tend to stretch and bulge out of shape. This stretching increases the likelihood of an aortic dissection, which is a tear or separation between the layers of tissue that make up the aorta. An aortic dissection usually causes severe pain in the abdomen, back, or chest, depending on the section of the aorta that is affected. Rupture of the aorta is a medical emergency requiring immediate surgery and medication.
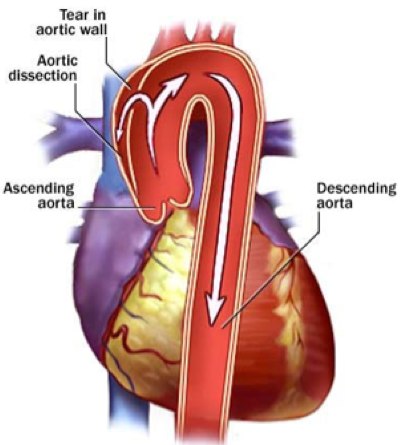
Dissection of the aorta can occur anywhere, may shear off side branches or leak into the heart sack (pericardium) and cause a condition called cardiac tamponade, all of which may be fatal if not treated promptly.
- Aortic regurgitation. A weakened and enlarged aorta may allow some blood to leak back into the heart during each heartbeat; this condition is called aortic regurgitation. Aortic regurgitation occasionally causes shortness of breath during normal activity. In serious cases, it causes the left ventricle of the heart to enlarge and may eventually lead to heart failure.
- Mitral valve prolapse. Between 75 and 85% of children with Marfan have loose or “floppy” mitral valves, which are the valves that separate the chambers of the heart. When these valves do not cover the opening between the chambers completely, the condition is called mitral valve prolapse. Complications of mitral valve prolapse include heart murmurs and arrhythmias. In rare cases, mitral valve prolapse can cause sudden death.
Diagnostic tests may include:
- Echocardiogram. This test is a sonogram of your heart. It uses sound waves to capture real-time images of your heart in motion. Echocardiograms show how well your heart chambers and valves are working. Occasionally, to better see your aorta, your doctor may recommend a transesophageal echocardiogram — in which the sound waves are generated from within your body by a device threaded down your esophagus.You’ll likely have an echocardiogram as the first test to check for Marfan syndrome. If your doctor thinks you have Marfan syndrome, the size of your aorta will be recorded, and your doctor will likely order another echocardiogram within six months to make sure your aorta hasn’t grown larger.
- Electrocardiogram (ECG or EKG). An ECG checks for heart rhythm problems, using adhesive electrodes attached to your chest.
- MRI or CT scans. These tests can help your doctor examine your aorta. MRIs use a strong magnet and radio waves to visualize soft tissues inside your body. A CT scan uses a special dye that can be seen on X-rays. The dye is injected into your vein, to produce images of your aorta.
Tests for blood relatives
Marfan syndrome and other conditions that affect the upper part of your aorta (thoracic aorta) tend to run in families. Because of this, your doctor may recommend that your first-degree relatives, such as your siblings or children, have tests to check for Marfan syndrome or other conditions. These tests include:
- Genetic testing. Marfan syndrome is a hereditary condition. Right now, there’s no genetic test that alone can definitely establish or rule out a diagnosis of Marfan syndrome. However, genetic testing can aid in the diagnosis of Marfan syndrome, especially if you don’t have symptoms. In addition, you may want to consider genetic testing and genetic counseling before starting a family, to see what your chances are of passing on Marfan syndrome to your future children.
Treatment
In the past, people with Marfan syndrome usually died of heart problems in their early 30s. Treatments to prevent aortic ruptures now allow many people with Marfan syndrome to live into their 70s. While no treatment exists for Marfan syndrome itself, therapy focuses on preventing the various complications of the disease. For that reason, the treatment you receive will depend on the nature and severity of your symptoms.
Cardiovascular problems
The cardiovascular complications association with Marfan syndrome can be life-threatening, so doctors typically recommend an annual heart exam.
Two main approaches exist for treating cardiovascular complications:
- Medications. Doctors often prescribe blood pressure lowering drugs to help prevent the aorta from enlarging and to reduce the risk of dissection, even though your blood pressure may be normal. The most commonly used drugs are beta blockers.
- Surgery. If your aorta’s diameter enlarges quickly or reaches a dangerous size — usually around 2 inches (5 cm) — your doctor may recommend an operation to replace a portion of your aorta with a tube made of synthetic material. This can help prevent a life-threatening rupture. Your aortic valve may need to be replaced as well. Some people with Marfan syndrome may require multiple operations.
